SARS-CoV-2 virus
3D model at
atomic resolution
SARS-CoV-2 virus
3D model at
atomic resolution
SARS-CoV-2 virus
3D model at
atomic resolution
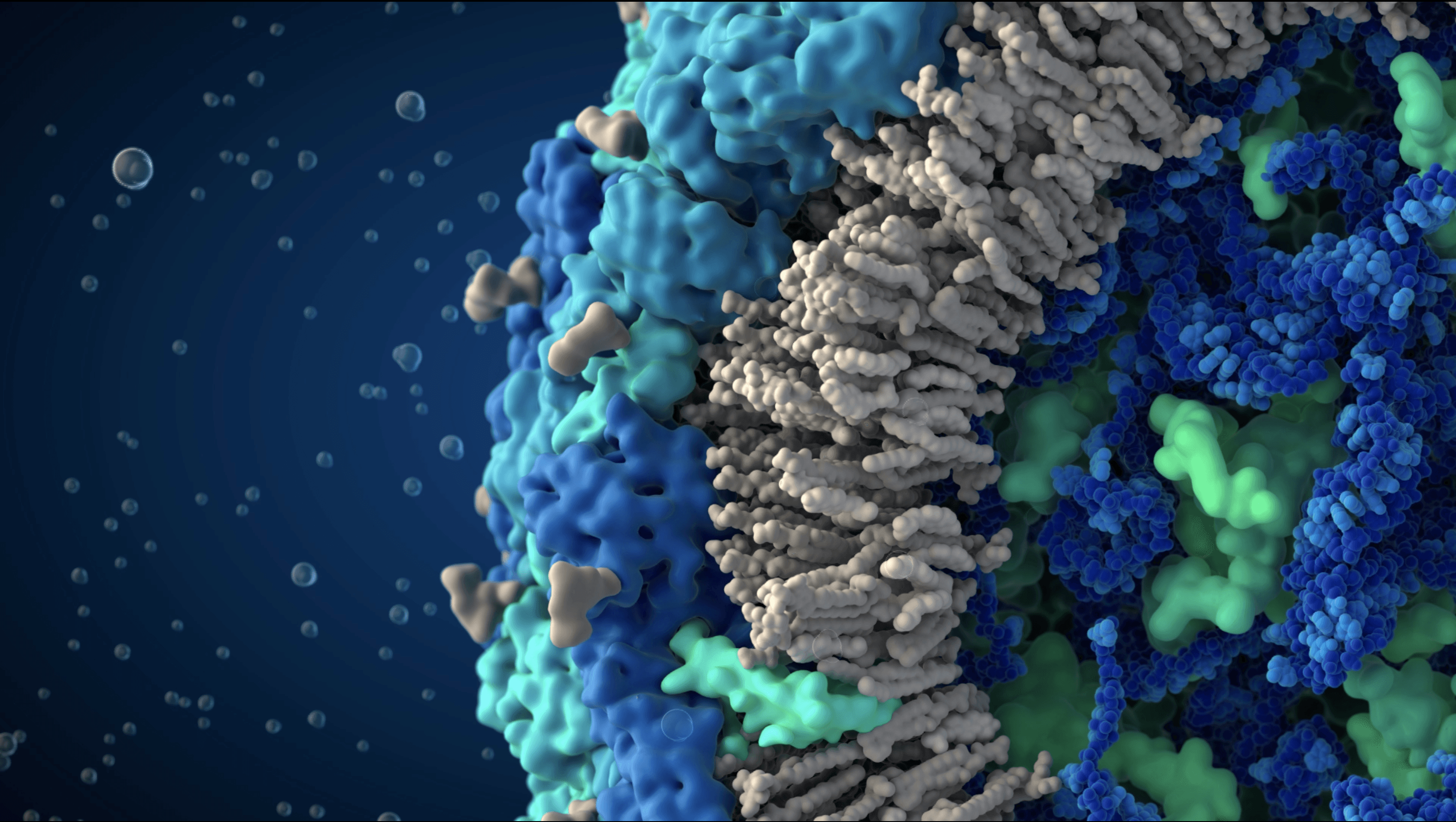
We created one of the most detailed and scientifically accurate atomic-resolution 3D models of the SARS-CoV-2 virus in 2020, based on the latest structural biology research and expert input from virologists. At the time of its release, the model represented the most accurate visualization of the SARS-CoV-2 viral particle available.
The project received broad international visibility shortly after publication. In May 2020, the model and accompanying visualization were featured by major science and medical news outlets, including EurekAlert!, Wiley Analytical Science, and News-Medical.net, highlighting its scientific accuracy and educational value. In 2021, the model was also featured in the CNN Special Report: The Origins of Covid-19 – Searching for the Source, bringing high-fidelity scientific visualization to a global audience during the COVID-19 pandemic.
We created one of the most detailed and scientifically accurate atomic-resolution 3D models of the SARS-CoV-2 virus in 2020, based on the latest structural biology research and expert input from virologists. At the time of its release, the model represented the most accurate visualization of the SARS-CoV-2 viral particle available.
The project received broad international visibility shortly after publication. In May 2020, the model and accompanying visualization were featured by major science and medical news outlets, including EurekAlert!, Wiley Analytical Science, and News-Medical.net, highlighting its scientific accuracy and educational value. In 2021, the model was also featured in the CNN Special Report: The Origins of Covid-19 – Searching for the Source, bringing high-fidelity scientific visualization to a global audience during the COVID-19 pandemic.
We created one of the most detailed and scientifically accurate atomic-resolution 3D models of the SARS-CoV-2 virus in 2020, based on the latest structural biology research and expert input from virologists. Developed using advanced structural bioinformatics techniques commonly applied in basic research and drug development, the model represented the most accurate visualization of the SARS-CoV-2 viral particle available at the time of its release.
The project received broad international visibility and media recognition shortly after publication. In May 2020, the model and accompanying visualization were featured by major science and medical news outlets, including EurekAlert!, Wiley Analytical Science, and News-Medical.net, highlighting its scientific accuracy and educational value. In 2021, the model was also featured in the CNN Special Report: The Origins of Covid‑19 – Searching for the Source, bringing high-fidelity scientific visualization to a global audience during the COVID-19 pandemic.
Project Goal
Project Goal
Project Goal
Project Goal
The goal of this project was to accurately represent the molecular structure of the SARS-CoV-2 virus at atomic resolution using current findings in structural biology and virology. The model was designed to support scientific understanding and education by providing a clear, research-driven visualization of viral architecture during a critical period of global public interest.
See full animation.
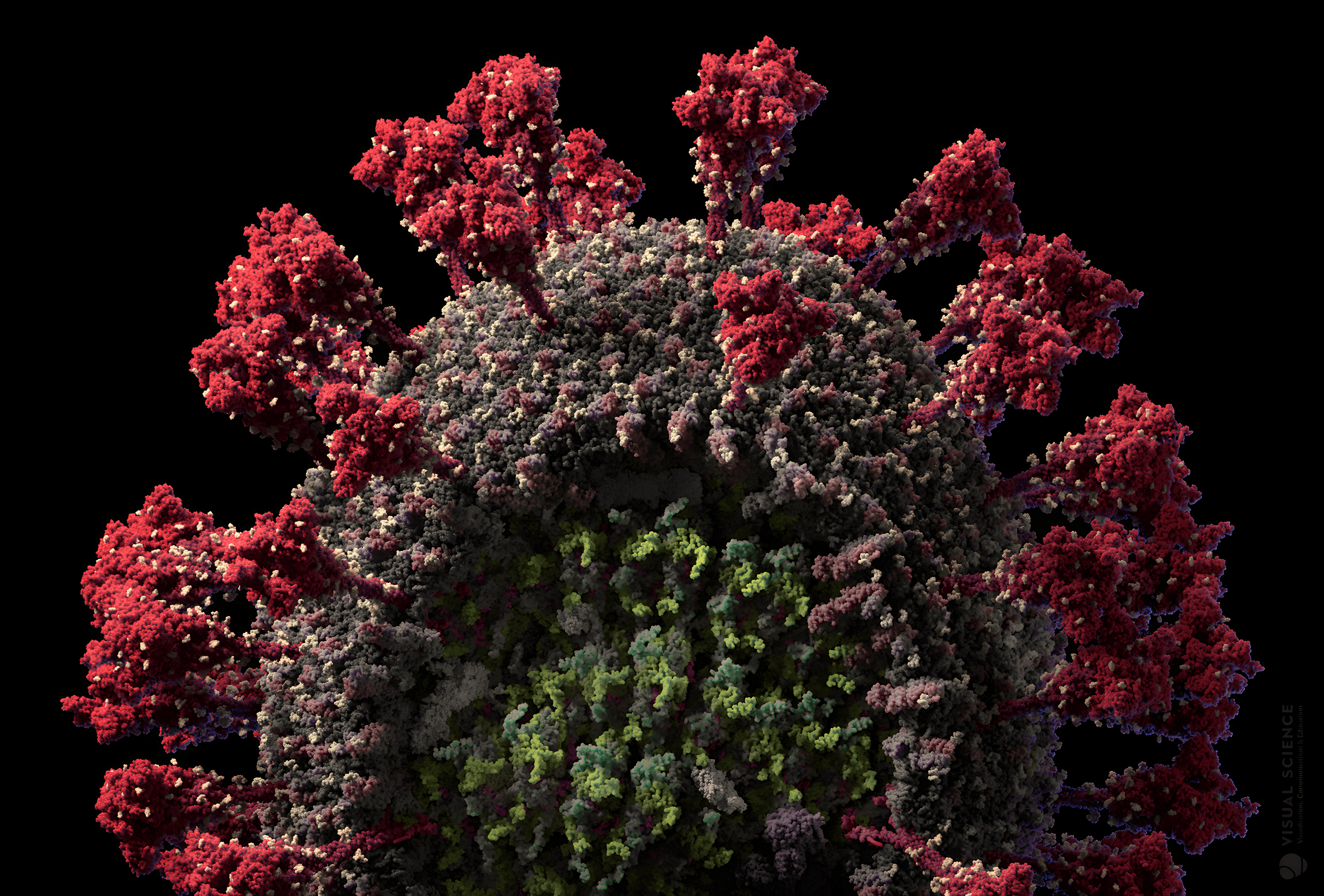
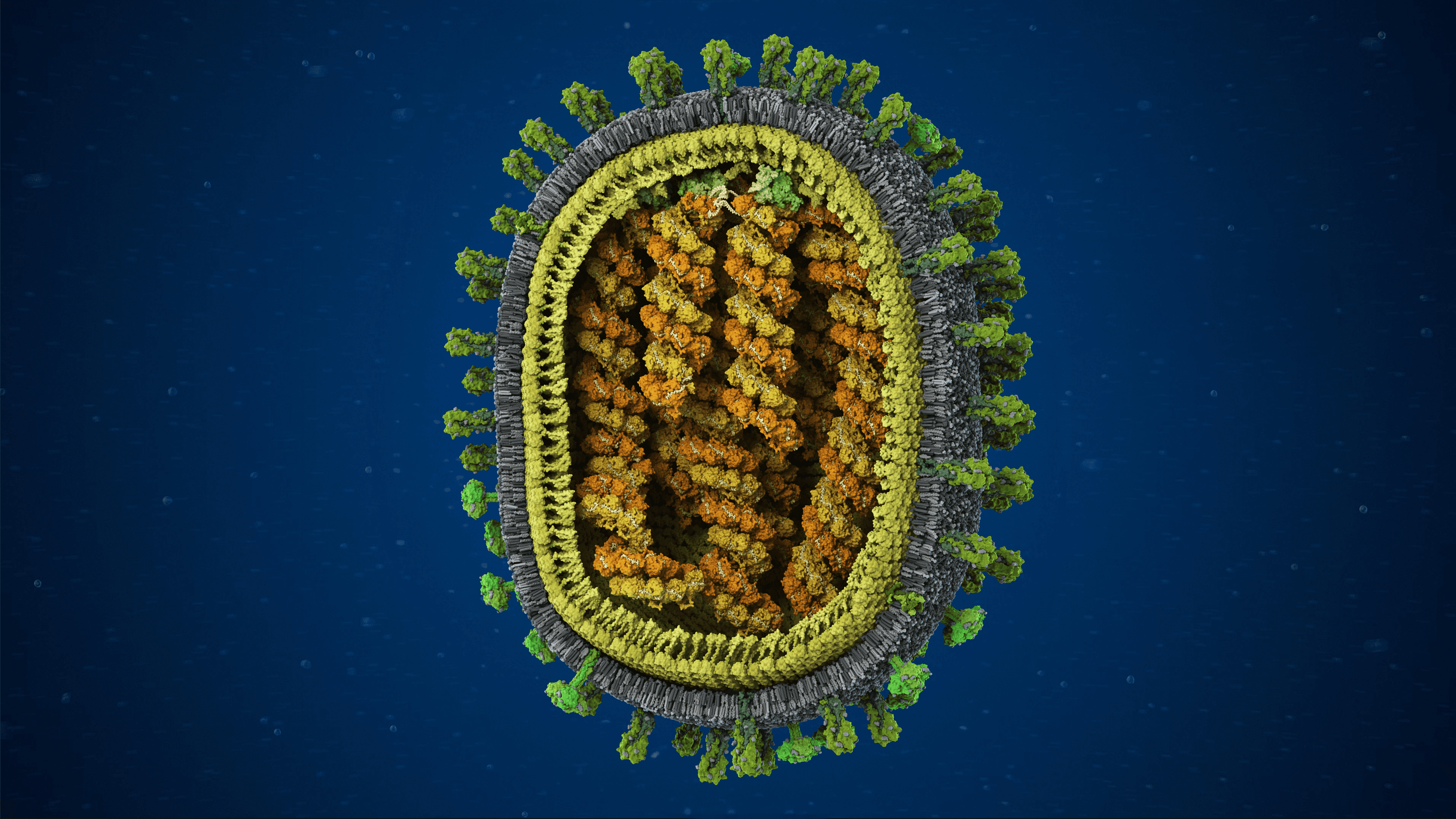
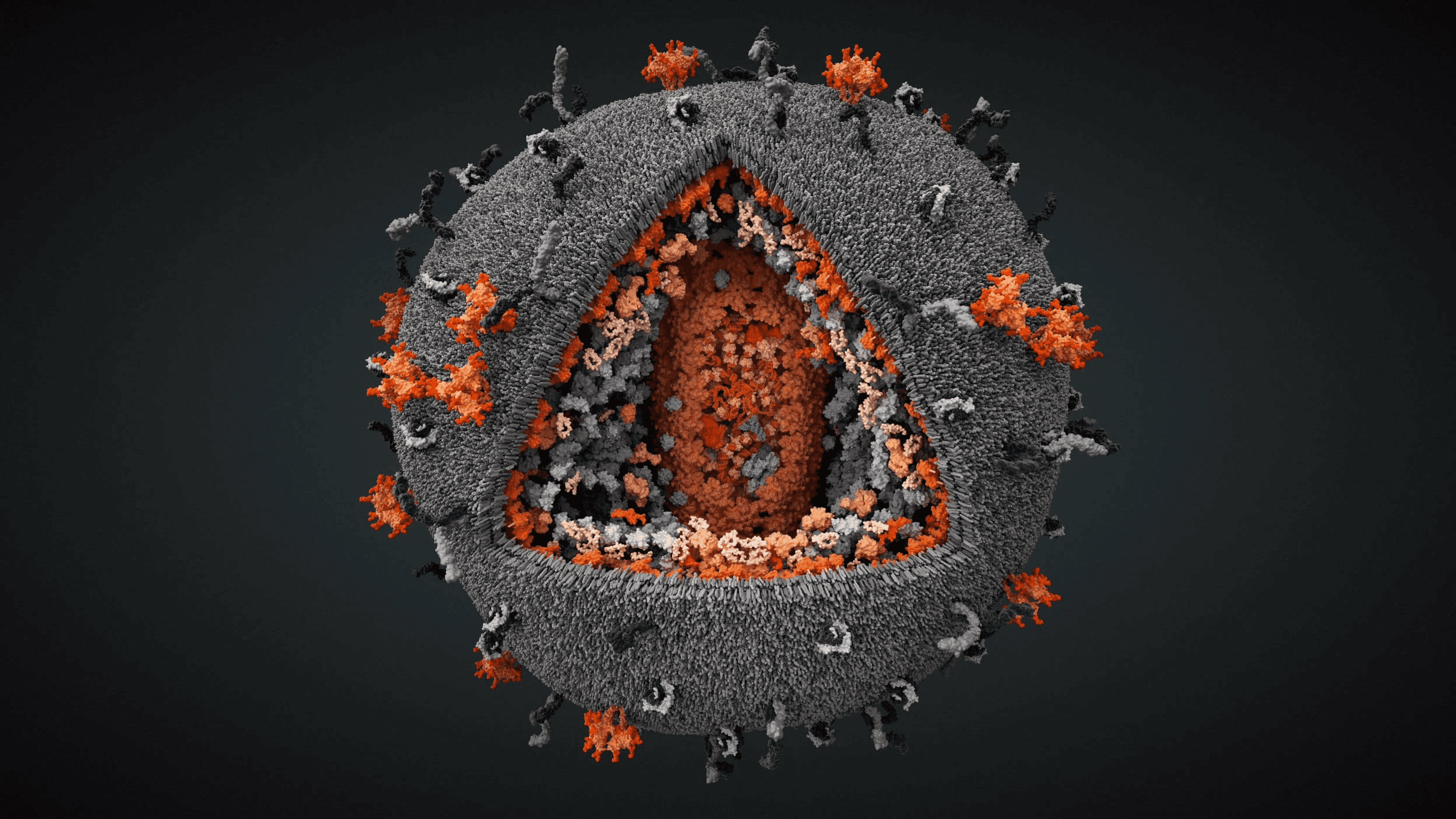
SARS-CoV-2 virus with exposed core
Virus core containing RNA and nucleocapsid protein
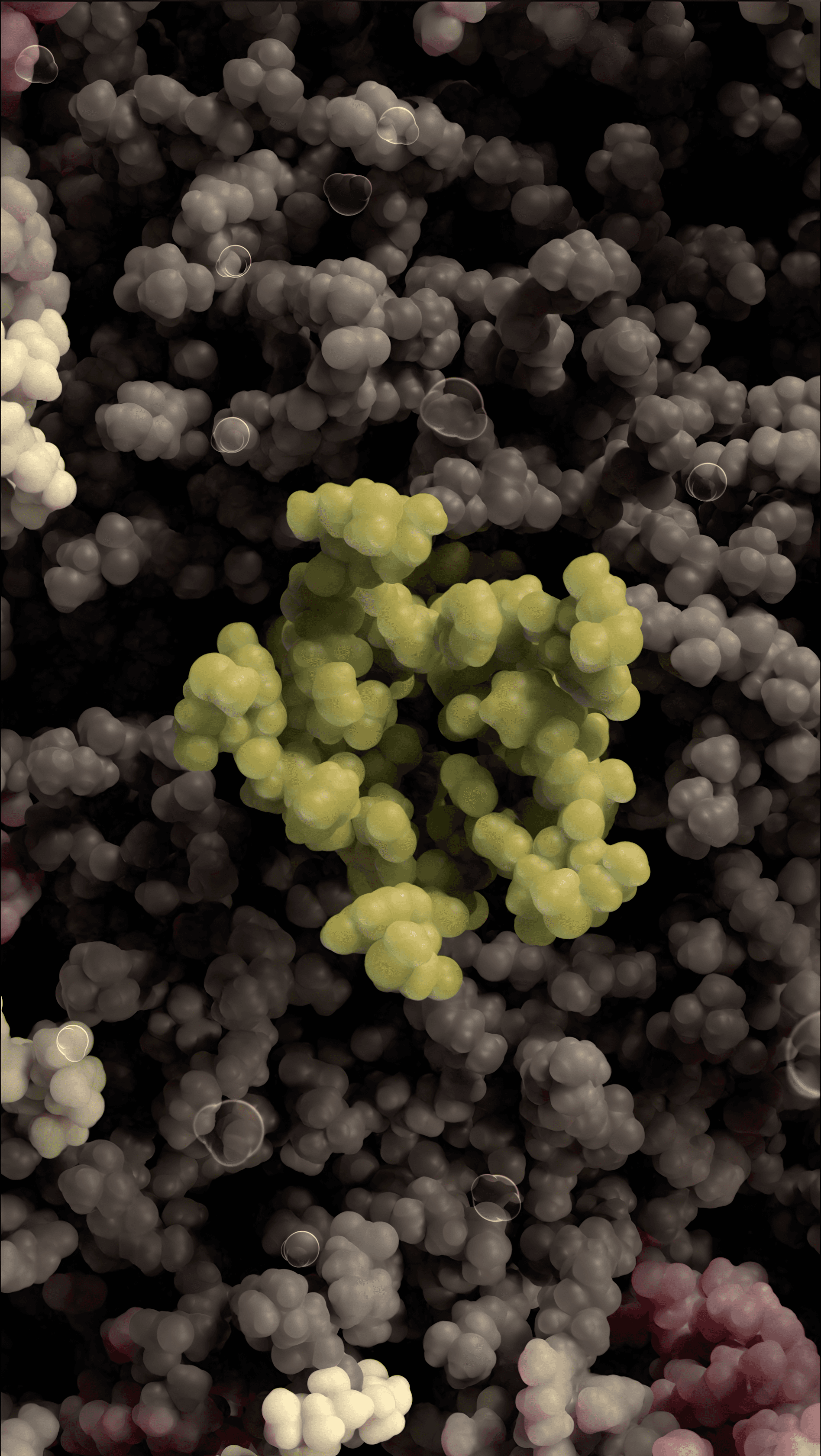
Envelope channel on membrane shell
“In their usual stunning style, the talented animators at Visual Science have created a model of the SARS-CoV-2 virus particle. These gorgeous visuals will enhance our understanding of the virus particle.”

Ph.D. Higgins Professor Department of Microbiology
and Immunology Columbia University
Ph.D. Higgins Professor Department of Microbiology
and Immunology Columbia University
“Visual Science created this highly detailed scientific 3D animation to illustrate the complexity of the SARS-CoV-2 virus particle. Spike proteins are shown on an unprecedented macro level.”
“Visual Science created this highly detailed scientific 3D animation to illustrate the complexity of the SARS-CoV-2 virus particle. Spike proteins are shown on an unprecedented macro level.”

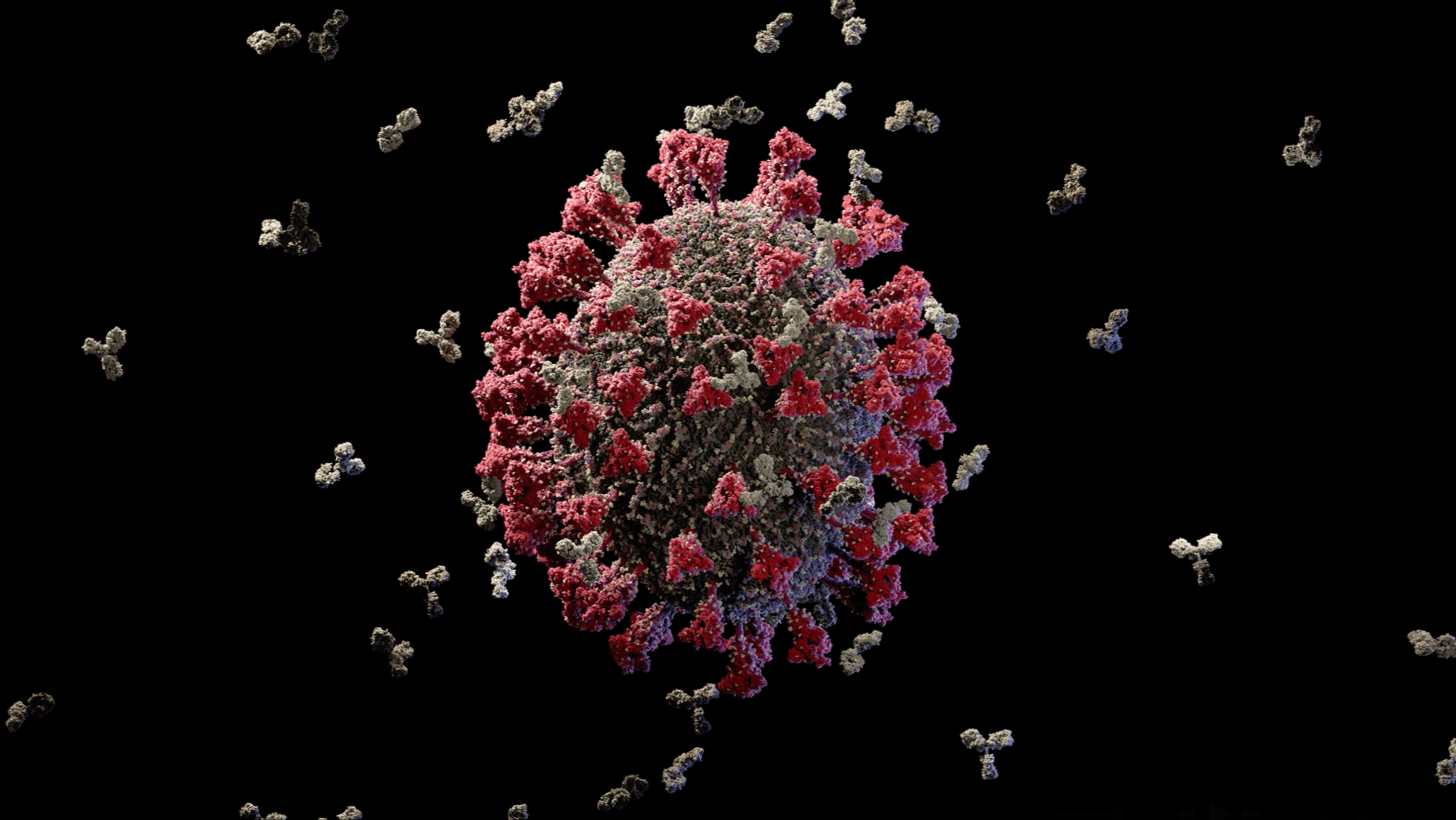
Immune Antibody’s attacking the virus
Immune Antibody’s attacking the virus
Immune Antibody’s attacking the virus
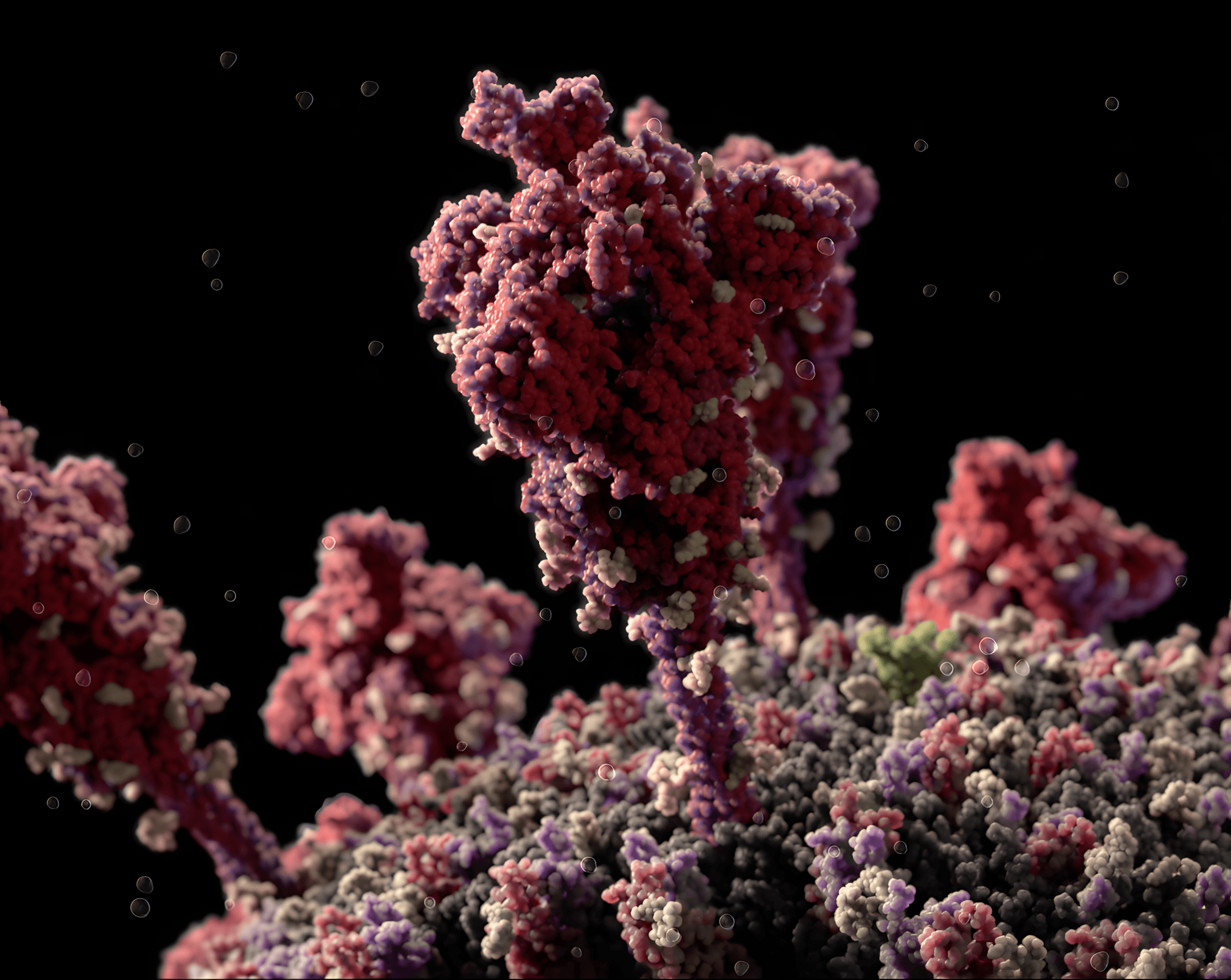
Protein spikes binding to host cell receptor
Protective Antibodies on host cell
The Process
The Process
Animation type:
Animation type:
High-End 3D
High-End 3D
Project timline:
Project timline:
5 weeks
5 weeks
Find out which of our 26 scientific animation options works best for investor relations and communications:
The SARS-CoV-2 model was developed using structural bioinformatics data derived from peer-reviewed scientific research and refined with input from expert virologists. Protein structures, lipid membranes, and host-derived components were integrated into a single, coherent viral particle model at atomic resolution, with all elements positioned according to known structural and biological constraints.
A consistent visual language was applied across the model, using color coding to distinguish viral proteins encoded by the viral genome from structures acquired from the host cell. This approach ensured clarity while maintaining fidelity to the biological data and supported both scientific interpretation and public understanding.
Why did we use this
animation type?
Why did we use this
animation type?
This project focuses on the highly complex molecular architecture of the SARS-CoV-2 virus, requiring our most advanced modeling approaches and a high-end 3D visualization style. High-end 3D molecular animation was essential to convey correct spatial relationships, scale, and organization of viral components that cannot be effectively communicated through conventional illustration.
Advanced control over geometry, materials, lighting, and camera movement made it possible to explore the viral particle from multiple perspectives while preserving scientific accuracy. Music and sound design were specifically composed for this animation to enhance its emotional impact and support the overall narrative. The visualization was designed to function as a concise and effective scientific narrative, uniting research-grade molecular precision with clear visual storytelling suitable for scientific, educational, and high-profile media use.
Outcome:
Outcome:
The completed SARS-CoV-2 model became a widely used and trusted visual resource for understanding viral structure at the molecular level. As part of Visual Science’s non-commercial Viral Park initiative, the project contributed to broader public and scientific engagement with virology and demonstrated the value of rigorous, high-fidelity molecular visualization for medical & scientific communication services during a global health crisis.
Full animation
Contact us
10/10

“
Fantastic team to partner with throughout the entire process. Excellent output and management.

Global Marketing Director, Pfizer
10/10
“
We are very pleased with Visual Science — they are a very responsive group to work with and the final product is exactly what we had envisioned.”

CEO, Atsena Therapeutics
10/10
“
The scientific expertise really showed in the discussions and final products. Everybody was responsive and great to work with, and the animations were both engaging and accurate!”

Program Manager, American Chemical Society
10/10
“
Visual Science team has just the right blend of scientific acumen and digital animation expertise to pull off these challenging projects.”

CEO, Apton Biosystems
9/10

“
Visual Science is a fantastic partner, capable of rendering the most complex science in compelling ways. They understood the science, and their production was excellent.”

VP, Head of Communications, Scorpion Tx
7/10

“
The structural biology models from Visual Science are stunning! Visual Science implemented our guidelines into beautiful scientific imagery for our website and scientific slide decks.”

Science Content Lead, Dewpoint Tx
9/10
“
I was very impressed with scientific knowledge of the Visual Science team, which led to a fantastic visualization of the MOA of our drug. Attention to scientific details was extraordinary.”

Vice President, R&D, Unicycive Tx
10/10
“
Fantastic team to partner with throughout the entire process. Excellent output and management.
Global Marketing Director, Pfizer
10/10
“
We are very pleased with Visual Science — they are a very responsive group to work with and the final product is exactly what we had envisioned.”
CEO, Atsena Therapeutics
10/10
“
The scientific expertise really showed in the discussions and final products. Everybody was responsive and great to work with, and the animations were both engaging and accurate!”
Program Manager, American Chemical Society
10/10
“
Visual Science team has just the right blend of scientific acumen and digital animation expertise to pull off these challenging projects.”
CEO, Apton Biosystems
9/10
“
Visual Science is a fantastic partner, capable of rendering the most complex science in compelling ways. They understood the science, and their production was excellent.”
VP, Head of Communications, Scorpion Tx
7/10
“
The structural biology models from Visual Science are stunning! Visual Science implemented our guidelines into beautiful scientific imagery for our website and scientific slide decks.”
Science Content Lead, Dewpoint Tx
9/10
“
I was very impressed with scientific knowledge of the Visual Science team, which led to a fantastic visualization of the MOA of our drug. Attention to scientific details was extraordinary.”
Vice President, R&D, Unicycive Tx
10/10
“
Fantastic team to partner with throughout the entire process. Excellent output and management.
Global Marketing Director, Pfizer
10/10
“
We are very pleased with Visual Science — they are a very responsive group to work with and the final product is exactly what we had envisioned.”
CEO, Atsena Therapeutics
10/10
“
The scientific expertise really showed in the discussions and final products. Everybody was responsive and great to work with, and the animations were both engaging and accurate!”
Program Manager, American Chemical Society
10/10
“
Visual Science team has just the right blend of scientific acumen and digital animation expertise to pull off these challenging projects.”
CEO, Apton Biosystems
9/10
“
Visual Science is a fantastic partner, capable of rendering the most complex science in compelling ways. They understood the science, and their production was excellent.”
VP, Head of Communications, Scorpion Tx
7/10
“
The structural biology models from Visual Science are stunning! Visual Science implemented our guidelines into beautiful scientific imagery for our website and scientific slide decks.”
Science Content Lead, Dewpoint Tx
9/10
“
I was very impressed with scientific knowledge of the Visual Science team, which led to a fantastic visualization of the MOA of our drug. Attention to scientific details was extraordinary.”
Vice President, R&D, Unicycive Tx
10/10
“
Fantastic team to partner with throughout the entire process. Excellent output and management.
Global Marketing Director, Pfizer
10/10
“
We are very pleased with Visual Science — they are a very responsive group to work with and the final product is exactly what we had envisioned.”
CEO, Atsena Therapeutics
10/10
“
The scientific expertise really showed in the discussions and final products. Everybody was responsive and great to work with, and the animations were both engaging and accurate!”
Program Manager, American Chemical Society
10/10
“
Visual Science team has just the right blend of scientific acumen and digital animation expertise to pull off these challenging projects.”
CEO, Apton Biosystems
9/10
“
Visual Science is a fantastic partner, capable of rendering the most complex science in compelling ways. They understood the science, and their production was excellent.”
VP, Head of Communications, Scorpion Tx
7/10
“
The structural biology models from Visual Science are stunning! Visual Science implemented our guidelines into beautiful scientific imagery for our website and scientific slide decks.”
Science Content Lead, Dewpoint Tx
9/10
“
I was very impressed with scientific knowledge of the Visual Science team, which led to a fantastic visualization of the MOA of our drug. Attention to scientific details was extraordinary.”
Vice President, R&D, Unicycive Tx

Contact us
10/10
“
Fantastic team to partner with throughout the entire process. Excellent output and management.
Global Marketing Director, Pfizer
10/10
“
We are very pleased with Visual Science — they are a very responsive group to work with and the final product is exactly what we had envisioned.”
CEO, Atsena Therapeutics
10/10
“
The scientific expertise really showed in the discussions and final products. Everybody was responsive and great to work with, and the animations were both engaging and accurate!”
Program Manager, American Chemical Society
10/10
“
Visual Science team has just the right blend of scientific acumen and digital animation expertise to pull off these challenging projects.”
CEO, Apton Biosystems
9/10
“
Visual Science is a fantastic partner, capable of rendering the most complex science in compelling ways. They understood the science, and their production was excellent.”
VP, Head of Communications, Scorpion Tx
7/10
“
The structural biology models from Visual Science are stunning! Visual Science implemented our guidelines into beautiful scientific imagery for our website and scientific slide decks.”
Science Content Lead, Dewpoint Tx
9/10
“
I was very impressed with scientific knowledge of the Visual Science team, which led to a fantastic visualization of the MOA of our drug. Attention to scientific details was extraordinary.”
Vice President, R&D, Unicycive Tx
10/10
“
Fantastic team to partner with throughout the entire process. Excellent output and management.
Global Marketing Director, Pfizer
10/10
“
We are very pleased with Visual Science — they are a very responsive group to work with and the final product is exactly what we had envisioned.”
CEO, Atsena Therapeutics
10/10
“
The scientific expertise really showed in the discussions and final products. Everybody was responsive and great to work with, and the animations were both engaging and accurate!”
Program Manager, American Chemical Society
10/10
“
Visual Science team has just the right blend of scientific acumen and digital animation expertise to pull off these challenging projects.”
CEO, Apton Biosystems
9/10
“
Visual Science is a fantastic partner, capable of rendering the most complex science in compelling ways. They understood the science, and their production was excellent.”
VP, Head of Communications, Scorpion Tx
7/10
“
The structural biology models from Visual Science are stunning! Visual Science implemented our guidelines into beautiful scientific imagery for our website and scientific slide decks.”
Science Content Lead, Dewpoint Tx
9/10
“
I was very impressed with scientific knowledge of the Visual Science team, which led to a fantastic visualization of the MOA of our drug. Attention to scientific details was extraordinary.”
Vice President, R&D, Unicycive Tx
10/10
“
Fantastic team to partner with throughout the entire process. Excellent output and management.
Global Marketing Director, Pfizer
10/10
“
We are very pleased with Visual Science — they are a very responsive group to work with and the final product is exactly what we had envisioned.”
CEO, Atsena Therapeutics
10/10
“
The scientific expertise really showed in the discussions and final products. Everybody was responsive and great to work with, and the animations were both engaging and accurate!”
Program Manager, American Chemical Society
10/10
“
Visual Science team has just the right blend of scientific acumen and digital animation expertise to pull off these challenging projects.”
CEO, Apton Biosystems
9/10
“
Visual Science is a fantastic partner, capable of rendering the most complex science in compelling ways. They understood the science, and their production was excellent.”
VP, Head of Communications, Scorpion Tx
7/10
“
The structural biology models from Visual Science are stunning! Visual Science implemented our guidelines into beautiful scientific imagery for our website and scientific slide decks.”
Science Content Lead, Dewpoint Tx
9/10
“
I was very impressed with scientific knowledge of the Visual Science team, which led to a fantastic visualization of the MOA of our drug. Attention to scientific details was extraordinary.”
Vice President, R&D, Unicycive Tx
10/10
“
Fantastic team to partner with throughout the entire process. Excellent output and management.
Global Marketing Director, Pfizer
10/10
“
We are very pleased with Visual Science — they are a very responsive group to work with and the final product is exactly what we had envisioned.”
CEO, Atsena Therapeutics
10/10
“
The scientific expertise really showed in the discussions and final products. Everybody was responsive and great to work with, and the animations were both engaging and accurate!”
Program Manager, American Chemical Society
10/10
“
Visual Science team has just the right blend of scientific acumen and digital animation expertise to pull off these challenging projects.”
CEO, Apton Biosystems
9/10
“
Visual Science is a fantastic partner, capable of rendering the most complex science in compelling ways. They understood the science, and their production was excellent.”
VP, Head of Communications, Scorpion Tx
7/10
“
The structural biology models from Visual Science are stunning! Visual Science implemented our guidelines into beautiful scientific imagery for our website and scientific slide decks.”
Science Content Lead, Dewpoint Tx
9/10
“
I was very impressed with scientific knowledge of the Visual Science team, which led to a fantastic visualization of the MOA of our drug. Attention to scientific details was extraordinary.”
Vice President, R&D, Unicycive Tx
Contact us
Contact us
Visual Science is an award-winning medical animation and digital scientific communications company, trusted by leading biotech and pharmaceutical organizations since 2007, including J&J, Pfizer, Novartis, Roche, Takeda, Gilead, AbbVie, and 100+ others.
We specialize in science-grade MoA and MoD videos, medical animation, and scientific storytelling, as well as digital and AI-driven solutions for Medical Affairs, marketing, corporate communications, and investor relations.


© 2026, Visual Science. All rights reserved.



Visual Science is an award-winning medical animation and digital scientific communications company, trusted by leading biotech and pharmaceutical organizations since 2007, including J&J, Pfizer, Novartis, Roche, Takeda, Gilead, AbbVie, and 100+ others.
We specialize in science-grade MoA and MoD videos, medical animation, and scientific storytelling, as well as digital and AI-driven solutions for Medical Affairs, marketing, corporate communications, and investor relations.
© 2026, Visual Science. All rights reserved.
Visual Science is an award-winning medical animation and digital scientific communications company, trusted by leading biotech and pharmaceutical organizations since 2007, including J&J, Pfizer, Novartis, Roche, Takeda, Gilead, AbbVie, and 100+ others.
We specialize in science-grade MoA and MoD videos, medical animation, and scientific storytelling, as well as digital and AI-driven solutions for Medical Affairs, marketing, corporate communications, and investor relations.
© 2026, Visual Science. All rights reserved.
Visual Science is an award-winning medical animation and digital scientific communications company, trusted by leading biotech and pharmaceutical organizations since 2007, including J&J, Pfizer, Novartis, Roche, Takeda, Gilead, AbbVie, and 100+ others.
We specialize in science-grade MoA and MoD videos, medical animation, and scientific storytelling, as well as digital and AI-driven solutions for Medical Affairs, marketing, corporate communications, and investor relations.
© 2026, Visual Science. All rights reserved.